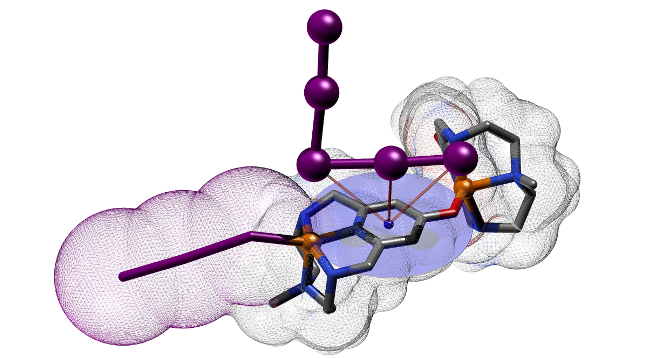
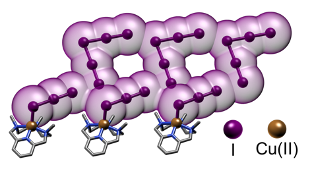
Abstract: Polyiodide networks are currently of great practical interest for the preparation of new electronic materials. The participation of metals in the formation of these networks is believed to improve their mechanical performance and thermal stability. Here we report the results on the construction of polyiodide networks obtained using Cu(II) complexes of a series of pyridinol-based tetraazacyclophanes as countercations. The assembly of these crystalline polyiodides takes place from aqueous solutions on the basis of similar structural elements, the [CuL]2+ and [Cu(H–1L)]+ (L = L2, L2-Me, L2-Me3) complex cations, so that the peculiarities induced by the increase of N-methylation of ligands, the structural variable of ligands, can be highlighted. First, solution equilibria involving ligands and complexes were analyzed (potentiometry, NMR, UV–vis, ITC). Then, the appropriate conditions could be selected to prepare polyiodides based on the above complex cations. Single-crystal XRD analysis showed that the coordination of pyridinol units to two metal ions is a prime feature of these ligands, leading to polymeric coordination chains of general formula {[Cu(H–1L)]}nn+ (L = L2-Me, L2-Me3). In the presence of the I–/I2 couple, the polymerization tendency stops with the formation of [(CuL)(CuH–1L)]3+ (L = L2-Me, L2-Me3) dimers which are surrounded by polyiodide networks. Moreover, coordination of the pyridinol group to two metal ions transforms the surface charge of the ring from negative to markedly positive, generating a suitable environment for the assembly of polyiodide anions, while N-methylation shifts the directional control of the assembly from H-bonds to I···I interactions. In fact, an extended concatenation of iodine atoms occurs around the complex dimeric cations, the supramolecular I···I interactions become shorter and shorter, fading into stronger forces dominated by the orbital overlap, which is promising for effective electronic materials.

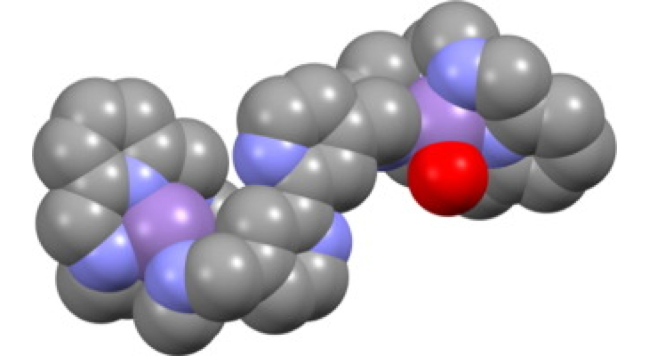
Abstract: Leishmaniasis is one of the world’s most neglected diseases with a worldwide prevalence of 12 million people. There are no effective human vaccines for its prevention, and outdated drugs hamper treatment. Therefore, research aimed at developing new therapeutic tools to fight leishmaniasis remains a crucial goal today. With this purpose in mind, here, we present 10 new compounds made up by linking alkylated ethylenediamine units to pyridine or quinoline heterocycles with promising in vitro and in 9 vivo efficacy against promastigote and amastigote forms of Leishmania infantum, Leishmania donovani, and Leishmania braziliensis species. Three compounds (2, 4, and 5) showed a selectivity index much higher in the amastigote form than the reference drug glucantime. These three derivatives affected the parasite infectivity rates; the result was lower parasite infectivity rates than glucantime tested at an IC25 dose. In addition, these derivatives were substantially more active against the three Leishmania species tested than glucantime. The mechanism of action of these compounds has been studied, showing alterations in glucose catabolism and leading to greater levels of iron superoxide dismutase inhibition. These molecules could be potential candidates for leishmaniasis chemotherapy due to their effectiveness and their ready synthesis.
Abstract: Nucleic acids are essential biomolecules in living systems and represent one of the main targets of chemists, biophysics, biologists, and nanotechnologists. New small molecules are continuously developed to target the duplex (ds) structure of DNA and, most recently, RNA to be used as therapeutics and/or biological tools. Stimuli-triggered systems can promote and hamper the interaction to biomolecules through external stimuli such as light and metal coordination. In this work, we report on the interaction with ds-DNA and ds-RNA of two aza-macrocycles able to coordinate Zn2+ metal ions and form binuclear complexes. The interaction of the aza-macrocycles and the Zn2+ metal complexes with duplex DNA and RNA was studied using UV thermal and fluorescence indicator displacement assays in combination with theoretical studies. Both ligands show a high affinity for ds-DNA/RNA and selectivity for ds-RNA. The ability to interact with these duplexes is blocked upon Zn2+ coordination, which was confirmed by the low variation in the melting temperature and poor displacement of the fluorescent dye from the ds-DNA/RNA. Cell viability assays show a decrease in the cytotoxicity of the metal complexes in comparison with the free ligands, which can be associated with the observed binding to the nucleic acids.
Abstract: Ordered polyiodide networks have recently gathered considerable attention as electronic materials, a field historically dominated by metals. Could we incorporate metal cations into polyiodide frameworks in a controlled manner to simultaneously boost electronic properties and robustness of these materials? Herein we present a first principle study featuring three analogous polyazacyclophanes (L, L-Me, L-Me3), differing only for the extent of N-methylation. We demonstrate (potentiometry, ITC) how they all form the same CuL2+ (L= L, L-Me, L-Me3) complex as prevalent species in solution, so that a level playing field exist where only N-methylation distinguishes them. Then we use them as countercation for polyiodide growth. XRD analysis on the resulting crystals clearly show that methylation is a valuable tool to gradually shift directional control of subtending pairing preferences from a H-bond to I···I interactions: this affects global packing and actively incorporates metal centres into polyiodide chains, setting the scene for further developments.
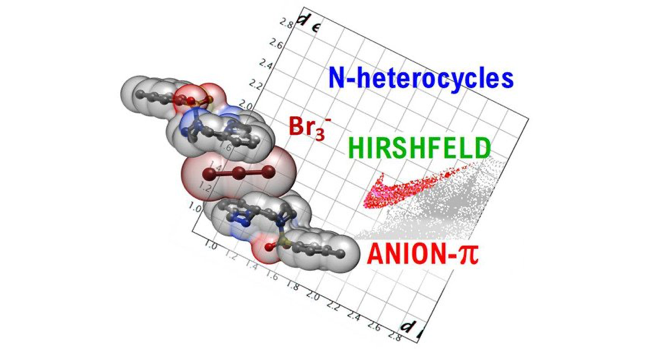
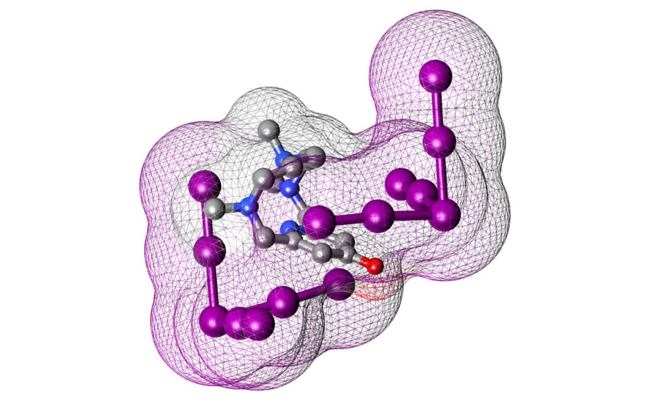
Abstract: Tetraaza-macrocyclic pyridinophane L-Ts, decorated with a p-toluenesulfonyl (tosyl; Ts) group, appear to be a useful tool to provide evidence on how the interplay of various supramolecular forces can help stabilise exotic anionic species such as tribromide (Br3−) anions. Indeed, crystals of (H2L-Ts)(Br3)1.5(NO3)0.5 unexpectedly grew from an acidic (HNO3) aqueous solution of L-Ts in the presence of Br- anions. The crystal structure of this compound was determined by single crystal XRD analysis. Hydrogen bonds, salt-bridges, anion-π, π-π stacking, and van der Waals interactions contribute to stabilising the crystal lattice. The observation of two independent Br3- anions stuck over the π-electron densities of pyridine and tosyl ligand groups, one of them being sandwiched between two pyridine rings, corroborates the significance of anion-π interactions for N-containing heterocycles. We show herein the possibility of detecting anion-π contacts from fingerprint plots generated by Hirshfeld surface analysis, demonstrating the effective usage of this structural investigation technique to further dissect individual contributions of stabilising supramolecular forces.
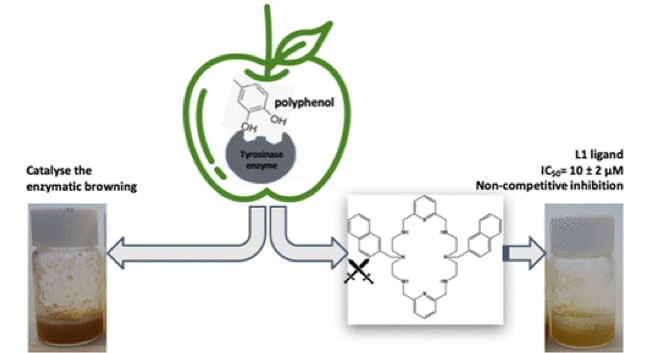
Abstract: Enzymatic browning is one of the main problems faced by the food industry due to the enzyme polyphenol oxidase (PPO) action provoking undesirable colour change in the presence of oxygen. Here, we report the evaluation of ten different azamacrocyclic compounds with diverse morphologies as potential inhibitors against the activity of PPO, both in model and real systems. An initial screening of ten ligands show that all aza- macrocyclic compounds inhibit to some extent the enzymatic browning, but the molecular structure plays a crucial role on the power of inhibition. Kinetic studies of the most active ligand (L2) reveal a S-parabolic I-parabolic non-competitive inhibition mechanism and a remarkable inhibition at micromolar concentration (IC50 = 10 μM). Furthermore, L2 action has been proved on apple juice significantly reducing the enzymatic browning.
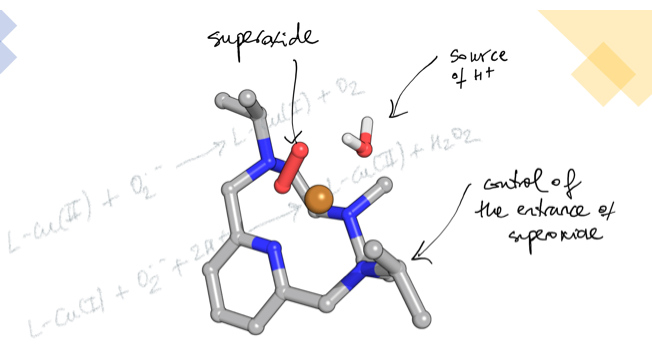
Abstract: Quantum chemical and multiscale calculations reveal the mechanistic pathway of two SOD mimetic N-alkylated tetra-azacyclophane copper complexes with remarkable activity. The arrangement of the binding side afforded by the bulky alkyl substituents and the coordinated water molecule as a proton source have key roles in the reaction mechanism.
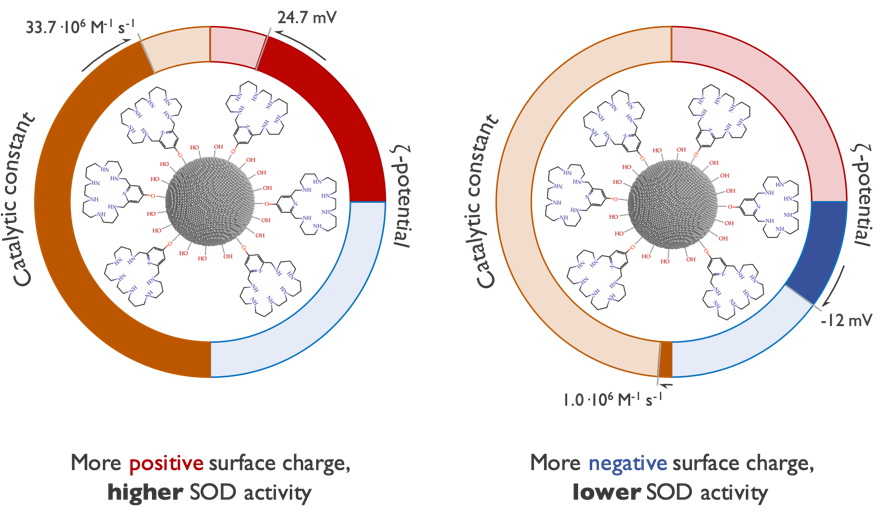
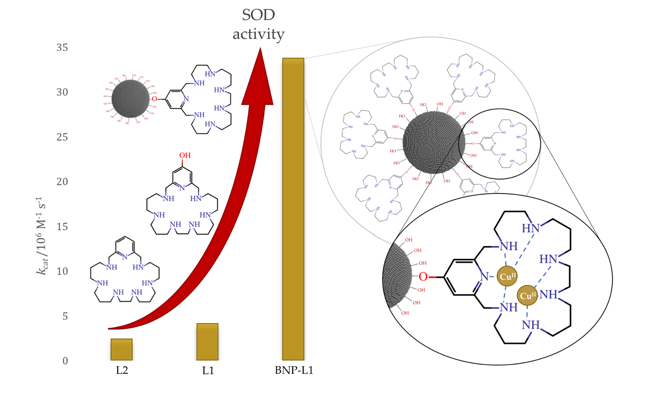
Abstract: The dismutation of superoxide radical anions is a key metabolic process to prevent oxidative damage. Inspired by the natural enzymes such as superoxide dismutases (SOD), we have designed a new set of aza-macrocycles, which form binuclear Cu2+ complexes, as shown by potentiometric and spectroscopic studies. The binuclear Cu2+ complexes show significant SOD activity, which is remarkably increased when the macrocycles are grafted onto boehmite nanoparticles (BNPs). The observed increase can be ascribed to the positive ζ-potential of the BNPs.
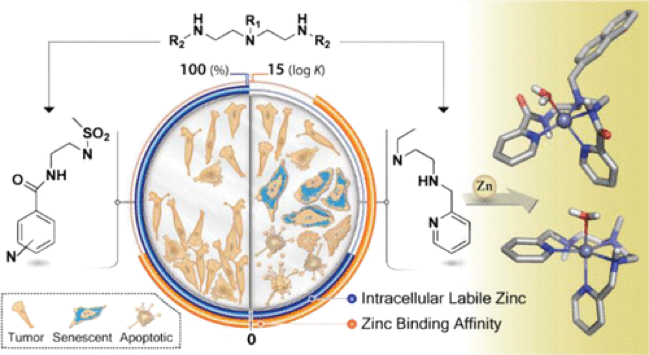
Abstract: In vitro viability assays against a representative panel of human cancer cell lines revealed that polyamines L1a and L5a displayed remarkable activity with IC50 values in the micromolar range. Preliminary research indicated that both compounds promoted G1 cell cycle arrest followed by cellular senescence and apoptosis. The induction of apoptotic cell death involved loss of mitochondrial outer membrane permeability and activation of caspases 3/7. Interestingly, L1a and L5a failed to activate cellular DNA damage response. The high intracellular zinc-chelating capacity of both compounds, deduced from the metal-specific Zinquin assay and ZnL2+ stability constant values in solution, strongly supports their cytotoxicity. These data along with quantum mechanical studies have enabled to establish a precise structure−activity relationship. Moreover, L1a and L5a showed appropriate drug-likeness by in silico methods. Based on these promising results, L1a and L5a should be considered a new class of zinc-chelating anticancer agents that deserves further development.
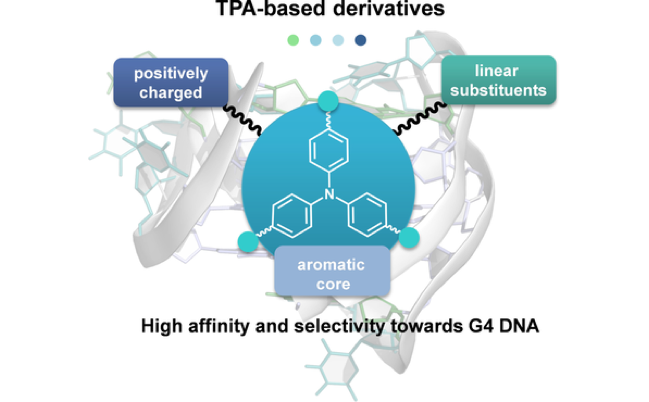
Abstract: Currently, significant efforts are devoted to designing small molecules able to bind selectively to guanine quadruplexes (G4s). These noncanonical DNA structures are implicated in various important biological processes and have been identi- fied as potential targets for drug development. Previously, a series of triphenylamine (TPA)-based compounds, including macrocyclic polyamines, that displayed high affinity towards G4 DNA were reported. Following this initial work, herein a series of second-generation compounds, in which the central TPA has been functionalised with flexible and adaptive linear polyamines, are presented with the aim of maximising the selectivity towards G4 DNA. The acid–base properties of the new derivatives have been studied by means of potentiometric titrations, UV/Vis and fluorescence emission spectroscopy. The interaction with G4s and duplex DNA has been explored by using FRET melting assays, fluorescence spectroscopy and circular dichroism. Compared with previous TPA derivatives with macrocyclic substituents, the new ligands reported herein retain the G4 affinity, but display two orders of magnitude higher selectivity for G4 versus duplex DNA; this is most likely due to the ability of the linear substituents to embrace the G4 structure.
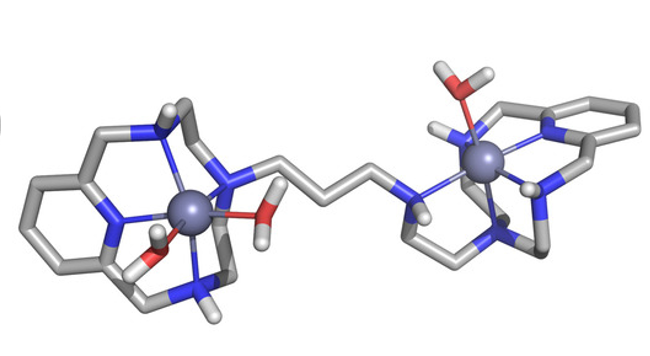
Abstract: Two binucleating hezaaza macrocycles containing a pyridinol spacer have been prepared and characterised. Protonation studies indicate the deprotonation of the phenol group at relatively low pH values with the concomitant occurrence of a keto-enolic equilibrium. These ligands readily form binuclear Cu2+ and Zn2+ complexes as denoted by potentiometric and spectroscopic studies. The binding of the metals yields to the ready deprotonation of the phenol with the stabilisation of the keto form that results in complexes of greater stabilities than the analogous ones containing pyridine as spacer instead of pyridine. Mixed Cu2+–Zn2+-complexes were also detected in aqueous solutions containing equimolar amounts of Cu2+, Zn2+ and ligands. The binuclear Cu complexes show significant SOD activity as proved by the McCord–Fridovich assays. The binuclear Cu2+ complexes of the ligands grafted to boehmite nanoparticles (BNPs) show a remarkable increase in SOD activity, which reaches 8-fold in one of the systems. The observed increase can be ascribed to the positive ζ-potential of the BNPs since the same complexes anchored to silica nanoparticles with negative z-potential do not show any apparent increase in activity. This behaviour is reminiscent of the positively charged funnel found in CuZnSOD, which has the electroactive copper ion at its end.
Abstract: Polyiodide chemistry is among the first historically reported examples of supramolecular forces at work. To date, owing to the increasingly recognized role of halogen bonding and the incorporation of iodine-based components in several devices, it remains an active field of theoretical and applied research. Herein we re-examine azacyclophanes as a class of ligands for the stabilization of iodine-dense three-dimensional networks, showing how we devised novel possible strategies starting from literature material. The new set of azacyclophane ligands affords novel crystal structures possessing intriguing properties, which develop on a double layer. At a macroscopic level, the obtained networks possess a very high iodine packing density (less than 2 times more diluted than crystalline I2): a simple parameter, IN, is also introduced to quickly measure and compare iodine packing density in different crystals. On the microscopic level, the present study provides evidence about the ability of one of the ligands to act as a three-dimensional supramolecular mold for the template synthesis of the rarely bserved heptaiodide (I7-) anion. Therefore, we believe our approach and strategy might be relevant for crystal engineering purposes.
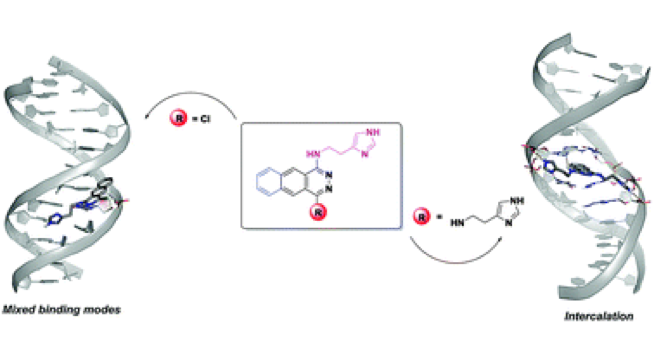
Abstract: The affinity and the binding mode of two benzo[g]phthalazine compounds, functionalized with one or two 2-(imidazole-4-yl)-ethylamine groups, to DNA and RNA models have been evaluated by means of UV-Vis, fluorescence and circular dichroism (CD) spectroscopies in combination with viscometry and molecular dynamics. Both organic molecules bind strongly to all nucleic acid models via the intercalation mode in the duplex structure, especially compound 1. Intriguingly, 1 exhibits different emission responses depending on the base composition of duplex DNA/RNAs, which points out the possibility of using it as a base selective nucleic acid probe. Moreover, the acid–base behaviour of both compounds has been studied by pH-metric titrations and UV-Vis and emission fluorescence techniques to investigate the unexplored basicity of this type of compound. 1 behaves as a triprotic base whilst 2 is a diprotic base with the protonation of the benzo[g]phthalazine moiety occurring in the physiological pH range.
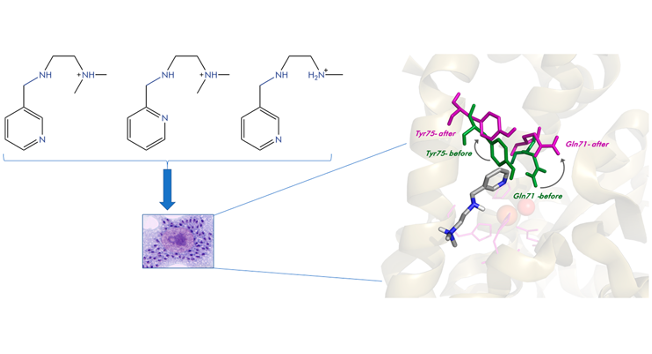
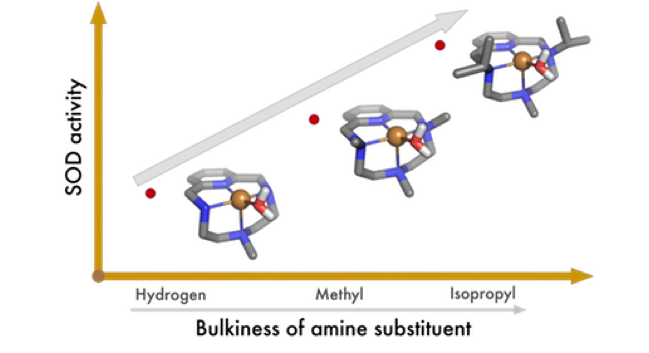
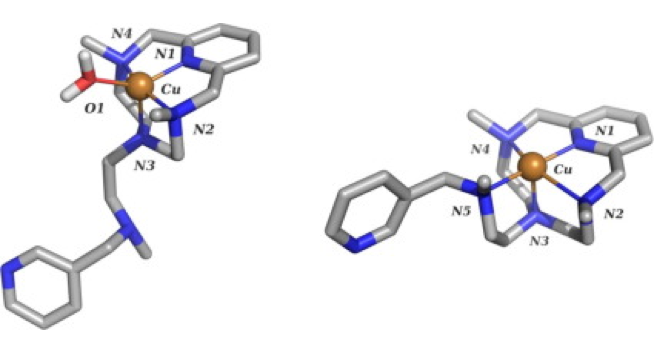
Abstract: A new tetraaza-pyridinophane macrocycle (L1) N-alkylated with two isopropyl and one methyl groups symmetrically disposed has been prepared and its behavior compared with those of the unsubstituted pyridinophane (L3) and the related compound with three methyl groups (L2). The protonation studies show that, first, a proton binds to the central methylated amine group of L1, while, second protonation leads to a reorganization of the protons that are at this stage attached to the lateral isopropylated amines. The X-ray structure of [HL1]+ agrees with the UV−vis and NMR studies as well as with the results of DFT calculations. The stability of the Cu2+ complexes decreases on increasing the bulkiness of the alkyl substituents of the amine groups. The crystal structures of [CuL1Cl](ClO4) and [CuL1(H2O)](ClO4)2·H2O show square pyramidal coordination geometries with the ligands disposed in a bent L-shaped conformation. Kinetic studies indicate that the rates of both complexation and ligand dissociation decrease with the bulkiness of the substituents, so that the stability changes are surely the results of compensating effects, complex formation dominating over complex dissociation. The pH dependence of the rate constants for complex formation cannot be explained by consideration of rapid pre-equilibria involving the different protonated forms of the ligand, and it has been interpreted in terms of a mechanism involving an acid−base equilibrium for a reaction intermediate. NBT SOD studies show that the Cu2+ complex of the bulkiest L1 ligand is the one having the highest activity (IC50 = 0.26(5) μM, kcat = 13.7 × 106 M-1 s-1) which can be associated with the poorer σ-donor ability of the tertiary amino groups, and the rigidity of the system, caused by the bulky isopropyl groups.
Abstract: The binuclear Cu2+ complex of a pyridinophane polyamine ligand ranking amongst the fastest SOD mimetics so far reported displays a remarkable SOD activity enhancement when grafted to the surface of boehmite (γ-AlO(OH)) nanoparticles (BNPs).
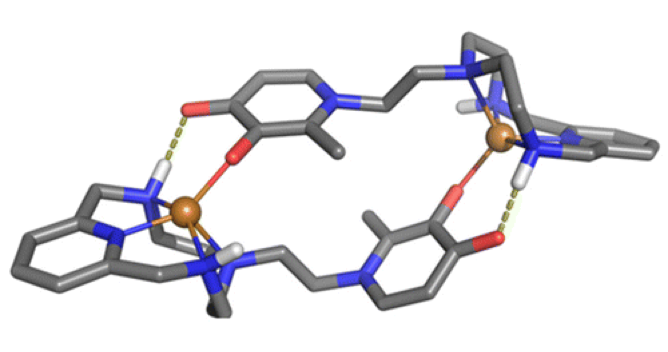
Abstract: Methylation of the secondary amine groups of a scorpiand-type ligand consisting of a pyridine spacer connected through methylene groups to a tris(2-aminomethyl) unit with the pendant arm further func- tionalised with a 3-pyridine unit leads to a ligand whose Cu(II) complex exhibits threefold enhanced SOD activity with respect to the non-methylated ligand. Potentiometric studies indicate the formation of [CuL]2+ species with a stability three orders of magnitude lower than that formed with the related non-methylated ligand. Kinetic studies indicate that methylation of the secondary nitrogens causes a deceleration of both the complex formation and the acid-induced dissociation of the metal ion. The reduction in stability associated to the poorer r-donor ability of the tertiary amino groups shifts the Cu(II)/Cu(I) redox potentials towards more positive values permitting a better cycling between both states needed for the dismutation of superoxide radicals.
Abstract: The synthesis, acid−base behavior, and Cu2+ coordination chemistry of a new ligand (L1) consisting of an azamacrocyclic core appended with a lateral chain containing a 3-hydroxy-2-methyl-4(1H)-pyridinone group have been studied by potentiometry, cyclic voltammetry, and NMR and UV−vis spectroscopy. UV−vis and NMR studies showed that phenolate group was protonated at the highest pH values [log K = 9.72(1)]. Potentiometric studies point out the formation of Cu2+ complexes of 1:2, 2:2, 4:3, 1:1, and 2:1 Cu2+/L1 stoichiometries. UV−vis analysis and electrochemical studies evidence the implication of the pyridinone moieties in the metal coordination of the 1:2 Cu2+/L1 complexes. L1 shows a stronger chelating ability than the reference chelating ligand deferiprone. While L1 shows no cytotoxicity in HeLa and ARPE-19 human cell lines (3.1−25.0 μg/mL), it has significant antioxidant activity, as denoted by TEAC assays at physiological pH. The addition of Cu2+ diminishes the antioxidant activity because of its coordination to the pyridinone moiety phenolic group.
Abstract: The Mn2+ coordination chemistry of double scorpiand ligands in which two polyazacyclophane macrocycles have been connected by pyridine, phenanthroline and bipyridine spacers has been studied by potentiometry, paramagnetic NMR and electrochemistry. All ligands show high stability with Mn2+ and the complexes were formed in a wide pH range. DFT calculations support the structures and coordination geometries derived from the study. A remarkable antioxidant activity was evidenced for these systems by the McCord-Fridovich assay and in Escherichia coli sodAsodB deficient bacterial cells. The three systems were tested as anti-inflammatory drugs in human macrophages measuring the accumulation of cytokines upon lipopolysaccharide (LPS) pro-inflammatory effect. All complexes showed anti-inflammatory effect, being [Mn2L1]4+ the most efficient one.
Copyright © 2020 - All Rights Reserved
Website by Àlvar Martínez-Camarena